Systemic autoinflammatory diseases are a group of conditions sharing a core of phenotypical similarities, as recently described by McGonagle, D. & McDermott, M. F. (PLoS Med. 3, e297 (2006)). They encompass several rare disorders characterized by extensive clinical and biological inflammation, and with no specific age or gender distribution in the human population. Although they were proposed to constitute a continuum of disorders with potential overlap, SAID should not be confused with the autoimmune family of diseases, related to adaptive immunity dysfunction and response to self-antigen(s). Primary physical manifestations of SAID classically involve fever, rash, joint pain or swelling, lymphadenopathy, and musculoskeletal symptoms in the absence of autoantibody response and microbial infection. It seems now well established that genetic mutations causing dysregulation of the innate immune system underlie the etiology of some SAID.
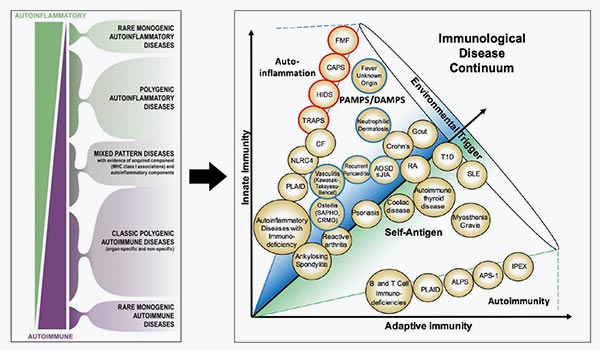
There is a continuum between autoinflammatory disorders, i.e., innate immunity dysfunction, and autoimmunity, i.e., adaptive immunity dysfunction. This concept has been proposed and schematized as follows by M McDermott et al, SAID can be roughly categorized into:
Monogenic familial disorders, in which genetic mutations have been identified (red circle in Figure )
Phenotype-based systemic inflammatory disorders, frequently sporadic, without identified mutation (blue circle): most of encountered syndromes belong to this category and constitute the biggest unmet need in terms of diagnosis. Evolving spectrum of systemic autoinflammatory diseases (SAIDs), affecting the innate immune system in comparison to autoimmune diseases that involve the adapted immune system, response to self-antigens and lymphocytes. Red circle, monogenetic SAID; blue circle, polyphenotype-based/ genetically undiagnosed SAID; PAMPS, pathogen-associated molecular patterns, DAMPs, danger-associated molecular pattern; ALPS, autoimmune lymphoproliferative syndrome; AOSD, adult onset Still’s disease; APS-1, autoimmune polyglandular syndrome type 1; CAPS, cryopyrin-associated periodic syndrome; CF, cystic fibrosis; CRMO, chronic recurrent multifocal osteomyelitis; FMF, familial mediterranean fever; HIDS, hyperimmunoglobulinemia D with periodic fever syndrome; IPEX, immune dysregulation polyendo-crinopathy enteropathy X-linked syndrome; RA, rheumatoid arthritis; sJIA, systemic juvenile idiopathic arthritis; SAPHO, synovitis-acne-pustulosis-hyperostosis-osteitis syndrome; SLE, systemic lupus erythematosus; T1D, type-1 diabetes; TRAPS, TNF receptor associated periodic syndrome. [Taken and adapted from McGonagle and McDermott, 2006 and Peckham et al, 2017].
Unmet needs
Due to the numerous symptoms observed in the different SAID-related conditions and their lack of specificity, diagnosis is challenging. Unlike autoimmune diseases whose autoantibodies are a tool for ascertaining the diagnosis, there is no known constitutive and disease-defining biomarker.
Diagnosis process is primarily clinical, and relies on the obtention of detailed patient’s history to fully understand the pattern of symptoms associated with disease flairs. The diagnostic difficulties posed by the SAID patients frequently lead to diagnostic delays and inadequate treatment decisions during the first weeks or months after SAID onset: for example, prescription of antibiotics under the presumption of severe systemic infection despite negative microbiologic investigation. On average, up to 5 inadequate or inefficient treatments are prescribed to each patient with SAID-related conditions before a correct diagnosis is made and initiation of adequate immunomodulating drugs. As SAID patients are seriously ill early after disease onset, such unsettled diagnosis procedure can have tremendous impact on patient’s health and quality of life.